Overview of PFIC and related diseases
Read on for an overall summary, or skip down for detailed information on specific types of PFIC.
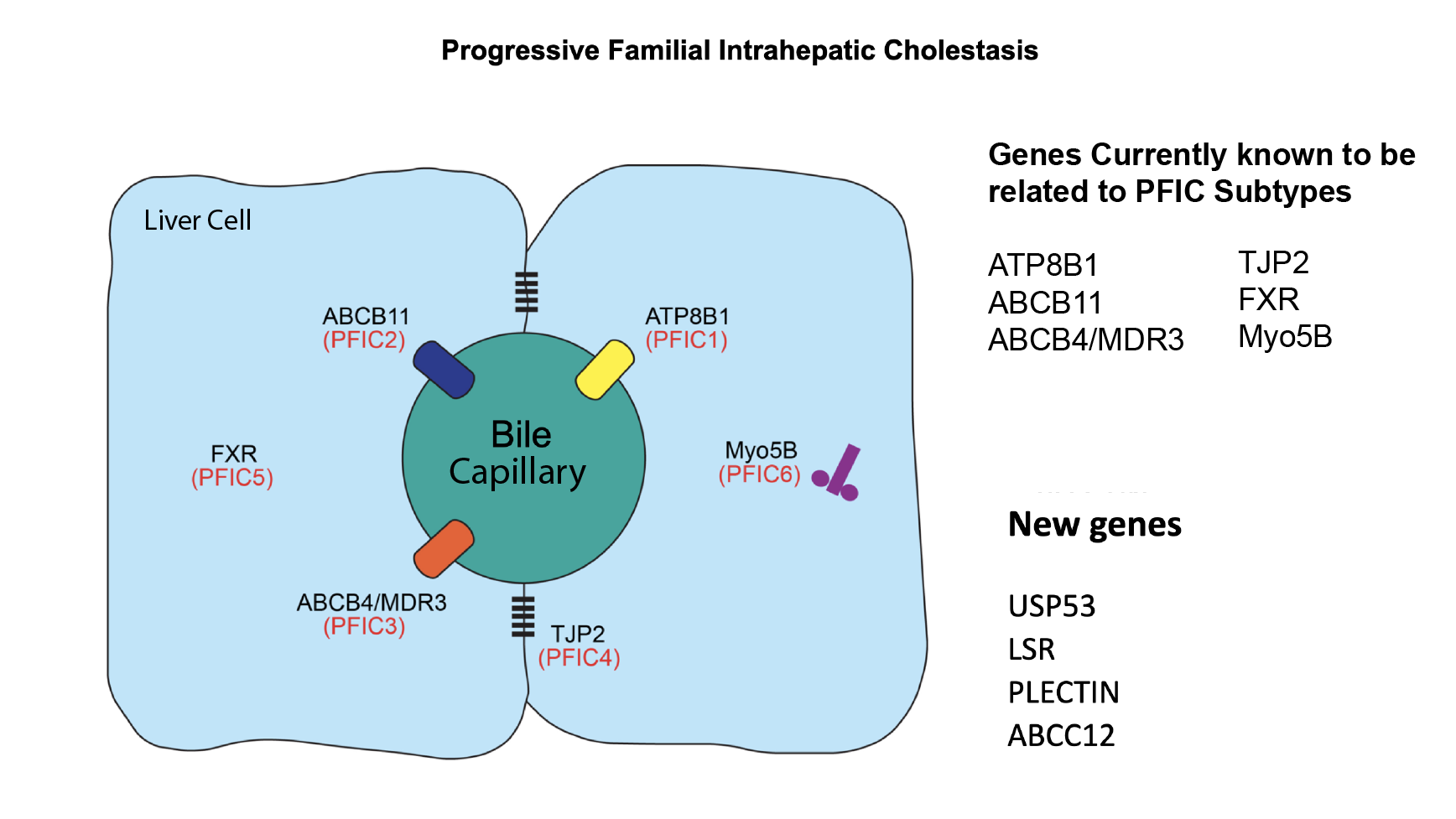
Progressive Familial Intrahepatic Cholestasis (PFIC) is an umbrella term for a group of rare genetic liver diseases that lead to cholestasis. Cholestasis is a failure of normal bile flow from the liver. While these conditions have similar symptoms, each PFIC type is caused by changes in a different gene. We now realize that these genes are associated with a range of different symptoms, and many people with mutations in these genes do not have the severe disease that was originally described. Scientists continue to discover new gene types that fall under the PFIC umbrella, helping us better understand the disease and personalize treatment.
| Disease | Affected Gene | Affected Protein |
|---|---|---|
| FIC1 deficiency (PFIC1) | ATP8B1 | FIC1 |
| BSEP deficiency (PFIC2) | ABCB11 | BSEP |
| MDR3 deficiency (PFIC3) | ABCB4 | MDR3 |
| TJP2 deficiency | TJP2 | TJP2 |
| FXR deficiency | NR1H4 | FXR |
| SLC51A deficiency | SLC51A | SLC51A/OSTa |
| Disease | Affected Gene | Affected Protein |
|---|---|---|
| USP53 deficiency | USP53 | USP53 |
| KIF12 deficiency | KIF12 | KIF12 |
| ZFYE19 deficiency | ZFYVE19 | ZFYE19 |
| MYO5B deficiency | MYO5B | Myosin 5B |
| SEMA7A deficiency | SEMA7A | SEMA7A |
| VPS33B deficiency | VPS33B | VPS33 |
*This table was updated in 2025 using information from the EASL Clinical Practice Guidelines on genetic cholestatic liver diseases Verkade, Henkjan J. et al. Journal of Hepatology, Volume 81, Issue 2, 303 – 325 and OMIM.org.
Genes are the code that we inherit that allow us to make different proteins. We all have approximately 20,000 genes; in most cases we inherit one copy of each gene from both of our parents. Changes in genes that reduce the function of the resulting protein are called mutations. The different types of PFIC involve mutations in different genes. In general a greater reduction in function is associated with more severe disease, but it is not always possible to predict. The proteins that are made using the PFIC-genes are all involved in bile production or flow. Deficiencies of these proteins cause bile build-up in the liver (“cholestasis”). For more information on the function of genes and proteins in relation to PFIC, please visit our genetics page.
A note on naming
You may see PFIC listed by number (1, 2, 3, etc.) or by its specific defective gene/protein. It’s important to understand that before any of these genes were identified, PFIC was all lumped together as low GGT and high GGT PFIC. As specific genetic etiologies were identified they started to be numbered. PFIC 1 2 and 3 were the first 3 identified and those terms stuck. As research progresses and we identify many more genetic causes of PFIC, the numbering system may stop being used. The term genetic cholestasis is becoming more commonly used to describe this group of diseases.
The journey to diagnosis can be hard.
Though genetic cholestasis typically appears in infancy or early childhood, some types, like MDR3 deficiency (PFIC3), may not present symptoms until adulthood.
Genetic cholestasis can also be missed in adults as the features can be very different from the original descriptions in children, and symptoms—like cholestatic pruritus (itch)—are often normalized or overlooked, adding to diagnosis challenges.
A breakdown of PFIC and related diseases
While many types of PFIC share key features and symptoms, each type also has its own distinct characteristics. Some types, such as FIC-1 deficiency and BSEP deficiency, have been recognized and studied for much longer than others. Because of this, clinicians and researchers have a better understanding of their typical symptoms, disease progression, and potential treatment options.
Some types of PFIC have only been discovered in recent years, thanks to advances in genetic testing and research. Much less is known about their natural history, prevalence, typical symptoms, and response to treatment compared to the more established types. As research continues and more patients are identified, our understanding of these newer types will grow.
Find Out More:
FIC-1 deficiency (PFIC 1)BSEP deficiency (PFIC 2)MDR3 Deficiency (PFIC 3)
TJP2 deficiency (PFIC 4)FXR Deficiency (PFIC5)MYO5B deficiencyBenign recurrent intrahepatic cholestasis (BRIC)
Unidentified Forms of PFIC
The mechanisms of bile formation are extremely complex. As a result, researchers anticipate they will identify more genetic causes of PFIC in the future. The identification of these possible new mutations requires state of the art genetic investigations. Although many PFIC patients have mutations in one of the genes mentioned above, there are still PFIC patients in whom mutations are not found in any of these genes. Genetic studies are underway to try to identify genetic factors contributing to PFIC in such patients.
FIC 1 deficiency (PFIC 1)
Previously, this type was known as Byler’s disease. PFIC1 is characterized by mutations in the ATP8B1 gene. These mutations cause a deficiency of the FIC1 protein. Deficiency of the FIC1 protein can lead to Benign Recurrent Intrahepatic Cholestasis (BRIC) as well as PFIC. These are now considered different levels of severity of the same condition, FIC1 deficiency.
It is not clear yet why exactly deficiency of the FIC1 protein causes build-up of bile in liver cells, and other symptoms of BRIC or PFIC.
Characteristics and symptoms of early onset FIC1 deficiency (PFIC1)
- Jaundice in the first months of life
- Severe itch (pruritus) beginning in the first year of life
- High levels of serum bile acids, low GGT levels
- Fat-soluble vitamin deficiencies are common (may require testing to identify)
- Progresses to chronic liver disease and possible cirrhosis at varying rates
- Potential problems that are independent of the liver, including hearing problems, irritation of the pancreas, chronic diarrhea, growth issues and chronic cough
BSEP deficiency (PFIC 2)
Mutations in the ABCB11 gene are responsible for the deficiency of the Bile Salt Export Pump (BSEP) protein. This protein works like a pump and moves bile salts out of liver cells. The deficiency of this protein directly leads to build-up of bile in liver cells, which in turn causes liver damage. Depending on the severity of the mutation, the BSEP protein is reduced or completely absent.
Characteristics and symptoms of early onset BSEP deficiency (PFIC 2)
- GGT levels are normal
- Pruritus (itch) becomes severe in the first years of life
- Cirrhosis (liver scarring) often occurs before the age of 10 years
- Milder intermittent forms (sometimes called BRIC) have also been identified
- There is a high risk of liver cancer in some individuals this type of PFIC which makes regular monitoring by blood test and ultrasound particularly important
MDR3 deficiency (PFIC 3)
Mutations in the ABCB4 gene cause deficiency of the MDR3 protein. This protein is involved in the transport of phospholipids (a type of fat) from liver cells into bile, where they bind to bile acids. Deficiency of this protein causes a lack of fats for bile acid to bind, which in turn causes injury to the tubes that drain bile from the liver. This can then lead to liver injury.
Characteristics and symptoms of early onset MDR3 deficiency (PFIC 3)
- Usually has high levels of GGT in the blood — this is an important difference from FIC1 and BSEP deficiencies
- Pruritus is not an early feature and tends to be milder
- Bile duct damage may occur
- Gallstones and stones within the liver are common
- There is a very wide range of severity
- MDR3 deficiency can occur during infancy, childhood or in adulthood – when it presents in childhood and young adulthood it may be with cirrhosis, an enlarged spleen or low platelets
- During pregnancy, women who are carriers for this disease can develop jaundice and itching
TJP2 deficiency (PFIC 4)
The TJP2 protein (Tight Junction Protein 2, sometimes called ZO2) plays a role in “tight junctions”. Tight junctions are areas where the membranes of two adjacent cells join to form a barrier. Such junctions are important throughout the body, and TJP2 is not specific to the liver. mild form of liver disease associated with mutations in the TPJ 2 gene was previously called familial hypercholanemia (high bile salts in blood). Episodic cholestasis has also been described. Only a small number of patients with TJP2 deficiency have been studied so far, so it is not yet understood what manifestations, other than liver disease and its consequences that TJP2 deficiency patients may have.
FXR Deficiency (PFIC 5)
This form of PFIC is caused by a mutation in the NR1H4 gene, which encodes the FXR (the Farsenoid X Receptor) protein. This protein is important in regulation of bile acid metabolism in the liver and intestine, as well as in other aspects of energy metabolism. Patients with PFIC due to FXR deficiency seem to develop rapidly progressing liver disease potentially very early in infancy. A very small number of patients have been reported so far, although it is expected more will likely be identified as this cause of PFIC is fairly newly described.
MYO5B deficiency
This is sometimes referred to as PFIC 10. MYO5B is involved in maintaining proper functioning of cell membranes and helping to move proteins, such as BSEP, to where they are needed. MY05B has been related to intestinal disorder, cholestasis, or both. Some patients with cholestasis due to MYO5B deficiency have progressive liver disease, while others have it only intermittently.
Benign recurrent intrahepatic cholestasis (BRIC)
BRIC stands for benign recurrent intrahepatic cholestasis. BRIC is not a separate type of PFIC, but rather an episodic presentation of some types of genetic cholestasis. Symptoms can last for weeks or months, but typically resolve and often do not lead to progression of liver disease, like PFIC, however proper diagnosis, monitoring of liver disease and symptom management are all just as important for patients who experience episodes of BRIC.
Characteristics and symptoms of episodic cholestasis (BRIC)
- Recurrent episodes of symptoms which may be quite mild
- First sign is often pruritus, but jaundice may also occur
- The time of a first episode varies widely (records show 2 months to 47 years)
- The first episode may be precipitated by factors such as infection or medication
- Frequency, duration, and severity of episodes can vary greatly
- Episodes often reduce with age
- In some cases a disease that starts with cholestatic episodes can become progressive; more like PFIC (i.e. there are no periods of remission of the symptoms) and can progress to chronic liver disease
Learn More: Educational Webinars
We offer several educational webinars that cover PFIC genetics in detail, especially in regards to how genetics and PFIC types and subtypes are related. Check out Genetics of PFIC and Its Subtypes (2020), Genetics of PFIC by Subtype (2021) and more!