Basic Overview of Genetics
Chromosomes are large molecules that mainly consist of DNA. Genes are short parts of a chromosome that contain the genetic code for heritable characteristics. Some characteristics such as height are influenced by many genes, and some just by one single gene. Humans have two copies for most genes including those genes associated with PFIC.
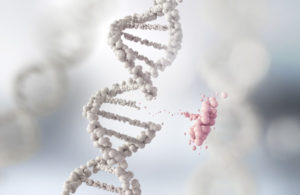
This image shows just one specific type of genetic mutation called a deletion. The white structure is the gene, the pink piece is the piece of gene that does not exist. This is just one type of mutation, there are many ways that a gene can be mutated.
A change in the genetic code is called a mutation. Mutations happen frequently. Some mutations are harmless, but some mutations can cause disease. The different subtypes of PFIC are due to mutations of a variety of specific single genes.
The mother and the father both have two copies of each gene, but they each only give one copy of their genes to the offspring/child. It is random which one of the two copies the child will get. So the child can have one of four different combinations. Suppose both parents have one healthy version (version 1) and one mutated version of the gene (version 2). The four possible combinations are:
- mother’s version 1 and father’s version 1 => 2 healthy copies
- mother’s version 1 and father’s version 2 => 1 healthy, 1 mutated copy
- mother’s version 2 and father’s version 1 => 1 healthy, 1 mutated copy
- mother’s version 2 and father’s version 2 => 2 mutated copies
Recessive vs. Dominant Disease
Some genetic diseases such as PFIC are due to mutations in just a single gene. The difference between recessive and dominant genetic diseases is whether one or two copies of the gene have to have a harmful mutation to actually show the disease.
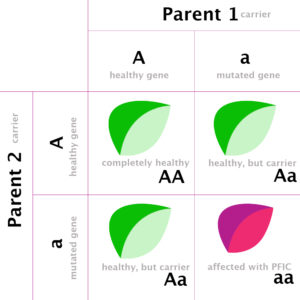
Note: Individuals with a single mutated gene are “carriers” of the recessive genetic disease: carriers are not sick themselves, but if they have a child with another carrier their children have a 1 in 4 chance of showing the disease (3 green logos = not sick, 1 purple logo = sick).
- Recessive diseases require two harmful copies of a gene to result in disease (see figure)
- Dominant disorders a single defective copy of the gene can lead to disease. The impact of that defective copy is dominant over the other copy which is healthy.
Here’s a bit more detail what the Figure tries to show:
Suppose both the father and the mother don’t have PFIC but both carry a mutation on one of their two copies of a gene related to a PFIC subtype (i.e. both parents are carriers, just as in the figure). There is a 50:50 chance that a sperm or egg will have a defective copy of the PFIC gene (sperms and eggs only have one copy of the gene). If both the egg and sperm have the defective copy of the PFIC gene, the resulting embryo (offspring) will have two copies of the defective PFIC gene and the offspring will likely have PFIC disease.
More generally, offspring have a 25% chance of being affected by the disease, while 50% of the offspring will be carriers and 25% will inherit two normal copies of the gene and be entirely unaffected. Note – if only one parent is a carrier and the other parent is unaffected, their offspring will have a 50% chance of being a carrier or 50% chance of being entirely unaffected.
Inheritance and Recessive vs Dominant
PFIC in general is a recessive disease and a person is only affected if she/he carries two mutated/harmful versions of the gene. However, some forms of PFIC can have features of either a dominant or recessive disease. For example, in certain circumstances of biological stress (e.g. pregnancy, infection, response to certain medications) individuals with only one affected copy of a recessive gene may develop symptoms of PFIC 3 (MDR3).
This video helps bridge the gap between Inheritance and Recessive vs Dominant. The content in this video is not PFIC specific but helps educate on the genetics behind the disease.
Credit for this video goes to Children’s National Hospital who posted it in 2019
This video helps bridge the gap between Inheritance and Recessive vs Dominant. The content in this video is not PFIC specific but helps educate on the genetics behind the disease.
The above information covers the very basics of genetic inheritance. Inheritance of a genetic disease such as PFIC is not that simple. Read on to take a closer look at genes, protein function, and the resulting severity of PFIC disease
A gene provides the code to build a specific protein. Different genes code for different proteins. Proteins regulate all kinds of processes in the body. A protein can have a very specific function. For example, let’s look at BSEP, which is the protein that is responsible for the disease PFIC 2. The function of BSEP is to transport bile acid out of the liver cell.
Mutations and partial/full loss of protein function
Remember that we have two copies of each gene. It is known that most PFIC subtypes are recessive, which means that in order to get sick both copies of the gene have to have a harmful mutation (see previous page). However, even if both copies have harmful mutations, the resulting disease can be more or less severe. It all depends on the type of mutation.
A gene can have different types of mutations, and individual patients can differ with respect to which particular mutation they have. Some mutations can still result in sufficient protein function. However, there are also mutations that result in partial or complete loss of protein function, leading to more severe disease. Let’s look at the BSEP protein again. Remember this is the protein responsible for disease in PFIC 2. Researchers have found different mutations in the gene that codes for BSEP. Only some of these mutations lead to a full loss of protein function, which generally leads to a more severe form of PFIC 2. You can read more about this latest research here.
Genetics of PFIC by Type
| Disease | Affected Gene | Affected Protein |
|---|---|---|
| FIC1 deficiency (PFIC1) | ATP8B1 | FIC1 |
| BSEP deficiency (PFIC2) | ABCB11 | BSEP |
| MDR3 deficiency (PFIC3) | ABCB4 | MDR3 |
| TJP2 deficiency | TJP2 | TJP2 |
| FXR deficiency | NR1H4 | FXR |
| SLC51A deficiency | SLC51A | SLC51A |
| Disease | Affected Gene | Affected Protein |
|---|---|---|
| USP53 deficiency | USP53 | USP53 |
| KIF12 deficiency | KIF12 | KIF12 |
| ZFYE19 deficiency | ZFYVE19 | ZFYE19 |
| MYO5B deficiency | MYO5B | Myosin 5B |
| SEMA7A deficiency | SEMA7A | SEMA7A |
| VPS33B deficiency | VPS33B | VPS33 |

Genetics of PFIC: Current Status and Implications
By Laura Bull, Ph.D. Professor Liver Center Laboratory, Department of Medicine, and Institute for Human Genetics University of California San Francisco
The attached paper was written to provide and overview of the current understanding of the genetics of PFIC. This paper was updated in 2018. She will be working on an update based on the most recent findings to be available in January, 2021.

Genetics of PFIC and its Subtypes
Join the PFIC Network and our host, Richard Thompson, Professor of Molecular Hepatology at the Institute for Liver Studies, Kings College London and Consultant Pediatric Hepatologist, at King’s College Hospital. He will discuss the seemingly complex topic of the Genetics of PFIC including the various subtypes and why they are each so unique. Dr. Thompson will also dive into some of the barriers behind genetic testing and why the disease is so difficult to treat. He will then give us a quick look at what has been recently discovered in the genetics of PFIC. Moderated by Executive Director, Emily Ventura, RN.